
El equipo argentino del Consorcio Internacional de Periodistas de Investigación (ICIJ) detectó al menos 13 episodios en los cuales grandes compañías extranjeras debieron retirar o corregir sus dispositivos medicos del mercado argentino porque se detectaron fallas con peligro de muerte para los pacientes, pero éstos tal vez nunca lo supieron. Algunos de los productos incluso ingresaron al país después de que saltaran las alertas en los Estados Unidos.
La información era hasta ahora “confidencial” según el Estado argentino. Pero la investigación periodística Implant Files logró que la Administración Nacional de Alimentos, Medicamentos y Tecnología Médica (ANMAT) admitiera parte de los datos que demuestran dos cosas: que las autoridades argentinas permitieron que ingresaran al país dispositivos médicos retirados del mercado en los Estados Unidos; y que las fabricantes e importadoras debieron retirar sus productos o corregirlos y que el organismo nunca lo comunicó a la población.
Los productos listados a continuación, son aquellos que la FDA al momento del alerta, identificó como Clase I, es decir peligrosos o defectuosos que podrían causar problemas de salud graves o incluso la muerte.
Dispositivos de recuperación Alligator
Uno de los productos de Medtronic que desató las alarmas en los Estados Unidos en 2014 es el Dispositivo de Recuperación Alligator, una pinza de platino diseñada para extraer cuerpos extraños de los vasos sensibles.
Es de un único uso. El problema detectado fue la deslaminación y desprendimiento del material de recubrimiento que podría esparcirse por el torrente sanguíneo del paciente.
Las autoridades sanitarias de los Estados Unidos informaron sobre las fallas en dos oportunidades: el 16 de abril de 2014 sobre 43 lotes y un segundo aviso el 9 de noviembre de 2016 sobre otros ocho modelos del Alligator.
“Existió una acción de campo”, se limitó a responder la ANMAT. El organismo asegura que el retiro o corrección del producto fue realizado por el importador. El alerta terminó recién el 11 de octubre de 2017.
En Buenos Aires, Medtronic brindó información específica sobre el producto. Admitió que el importador debió retirarlos de las instituciones médicas y destruirlos. “Todas las unidades que ingresaron a Argentina y que estaban afectadas por la alerta de la FDA fueron importadas y vendidas directamente por Biosud SA, uno de nuestros distribuidores. El 7 de octubre de 2016 –un mes antes de la publicación de la alerta en los Estados Unidos-, Biosud notificó voluntariamente a la ANMAT sobre esta alerta y se comunicó con las instituciones de atención médica que habían adquirido el producto para solicitarles que devolvieran el producto (a Biosud). Luego de completar el retiro de mercado, y alineado con los procedimientos para estos casos, los productos devueltos a Biosud fueron destruidos. El distribuidor proporcionó evidencia de la destrucción el día 14 de febrero de 2017 a Medtronic y a la ANMAT”.
Alambres guías orientables de Medtronic Vascular
Este pequeño dispositivo es utilizado para colocar un catéter y permitir el mejor acceso al corazón del paciente para un mejor contacto con los tejidos durante la cirugía. Se pueden usar para llegar a una lesión pero no están diseñados para la vasculatura cerebral.
El 15 de noviembre de 2013, la FDA de los Estados Unidos emitió una alerta luego de que Medtronic le informara al organismo de control que varios de los modelos de estos alambres sufrían un desperfecto: el teflón utilizado en la fabricación se “descaramaba”, como las espinas de un pescado. El desperfecto podía causas la muerte de los pacientes.
Para entonces, la ANMAT ya había autorizado la importación del producto a Medtronic Latin America en junio de 2012. El permiso incluía varios de los modelos que debieron ser retirados del mercado en los Estados Unidos: Zinger, Cougar Nitinol y Attain Hybrid.
El alerta cesó 15 de diciembre de 2014. Entre 2012 y 2018 fueron importados más de 2.000 unidades, incluso mientras la alarma estuvo vigente.
La ANMAT sostiene que el importador “retiró el producto del mercado” argentino.
Tras la consulta a Medtronic, se informó que “del total, sólo 40 unidades de este producto importadas a la Argentina resultaron afectadas por este recall”. De las 40 unidades, 34 fueron devueltas a la empresa el 21 de noviembre de 2013 y se destruyeron. Para cuando llegó la notificación de la FDA, las otras 6 unidades ya habían sido utilizadas.
Y añadieron: “No se recibieron observaciones, reclamos o problemas por parte de las instituciones de salud que las utilizaron”.
Medtronic es un gigante estadounidense. En el transcurso de este año, 252 periodistas y especialistas en datos de 59 organizaciones de noticias en 36 países entrevistaron o revisaron testimonios de más de 50 empleados de Medtronic y hablaron con docenas de funcionarios gubernamentales, pacientes, médicos y expertos. Los periodistas examinaron decenas de miles de páginas de registros judiciales, registros de empresas, informes regulatorios, auditorías gubernamentales, registros de cabildeo, transcripciones de llamadas de analistas, artículos de revistas médicas y otros documentos.
Esta compañía es uno de los modelos típicos del mercado de dispositivos médicos ahora cuestionado por esta investigación.
Charles Rosen es un cirujano de California y especialista en columna vertebral y en seguridad. Cofundó la Asociación para la Ética Médica para la defensa del paciente y sostuvo en una entrevista con ICIJ que el “Modelo Medtronic” consiste en desarrollar un producto, sacarlo rápido, oculta las complicaciones, pagar millones a consultores por estudios favorables, pagar por documentos para usar el dispositivo en pacientes que obtienen poco o ningún beneficio de ellos y facturar millones.
Luego, dice Rosen, la empresa "tiene que retirar el producto” del mercado por las fallas. Las ganancias de las ventas “se convierte en un ingreso para pagar los juicios" que le iniciarán los pacientes por los desperfectos que podrían haberles costado la vida.
El director ejecutivo de Medtronic, Omar Ishrak, no quiso hacer comentarios. En una declaración escrita, el vocero Rob Clark dijo que la compañía considera la seguridad del paciente como su principal prioridad y respalda los "estándares más altos de práctica ética"
Clark, negó que a los médicos se les pague por usar o promocionar productos de Medtronic. La compañía ha enfrentado investigaciones de corrupción en Brasil, India, China y Estados Unidos. Medtronic ha pagado al gobierno de los Estados Unidos y a cinco estados multas por US$ 127 millones para resolver seis casos de supuestos sobornos y otras prácticas fraudulentas desde 2008.
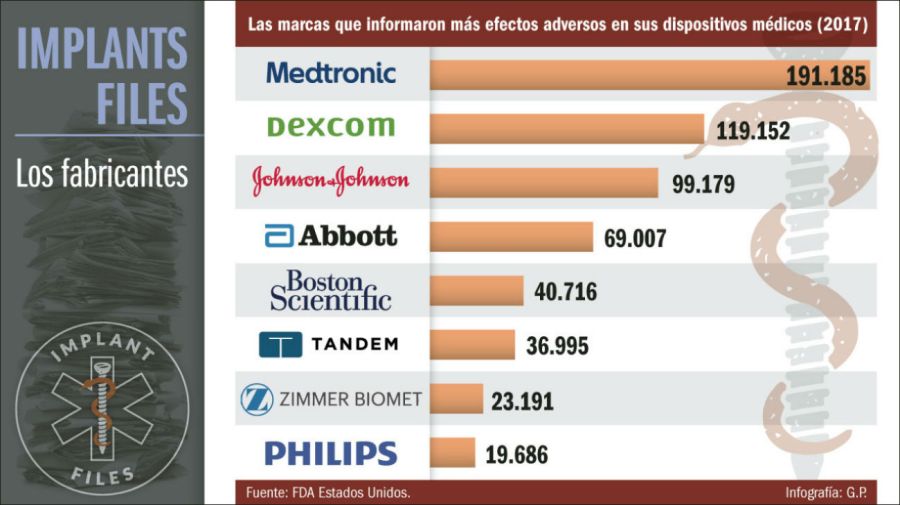
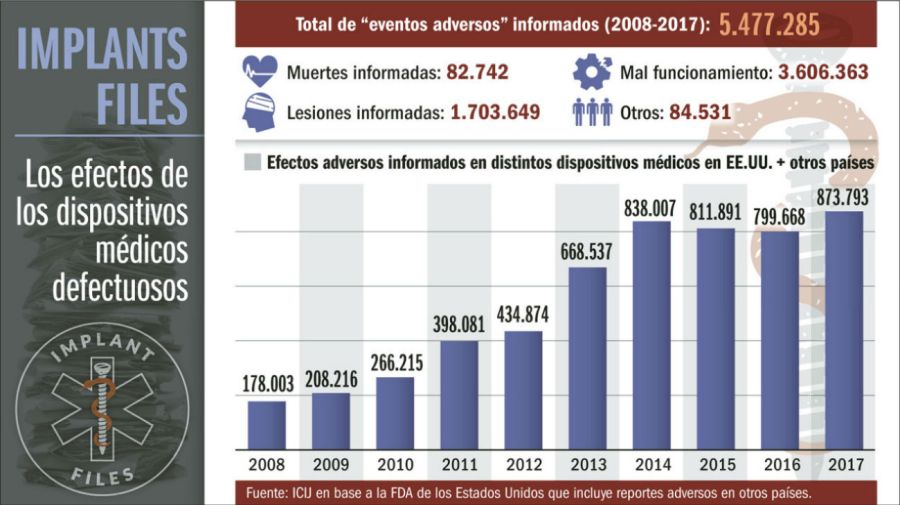
Las lagunas e inconsistencias en el sistema de dispositivos médicos son aprovechadas por la industria. Un análisis de ICIJ de los informes de eventos adversos presentados ante los reguladores de los Estados Unidos revela que entre 2008 y 2017 existieron reportes de 9.300 muertes y 292.000 lesiones potencialmente relacionadas con productos fabricados por Medtronic o sus subsidiarias.
“Medtronic identifica lagunas legales en el sistema y parece que las usa tanto o más que cualquier fabricante de dispositivos. Emplea revisiones aceleradas de sus productos, superando rigurosas pruebas científicas. Una revisión preliminar muestra que Medtronic gasta más en lobby que sus rivales y distribuye más efectivo para ganarse el favor de los médicos y pacientes”, revela ICIJ.
La compañía dice que planea resolver alrededor de 6.000 reclamos de personas presuntamente dañadas por su producto Infuse Spine y 12.000 reclamos relacionados con una malla pélvica. En Canadá, 9.000 personas están demandando debido a las lesiones de los cables del desfibrilador Sprint Fidelis de Medtronic.
Durante un período de tres años que finaliza en 2015 (la última información disponible), Medtronic y sus subsidiarias pagaron $ 186 millones en regalías, honorarios y otra remuneración a más de 57.000 médicos. Medtronic también dona dinero a grupos de defensa del consumidor como la American Heart Association (US$ 758.000) y Juvenile Diabetes Research Foundation (US$ 356.000) y a sociedades profesionales como Cardiovascular Research Society (US$ 5,1 millones) y el Congreso de Cirujanos Neurológicos (US$ 1,2 millones).
Otra de las compañías estadounidenses con alertas por fallas en sus dispositivos médicos es General Electric, que fabrica estos productos a través de su división GE Healthcare.
Esta compañía tuvo al menos tres tipos de dispositivos médicos que podrían causar la muerte de pacientes y que ingresaron a la Argentina. El equipo argentino identificó un sistema de resonancia magnética, una cámara de rayos gamma y un dispositivo de medicina nuclear denominado Infinia Hawkaye. Todos son equipos de grandes volúmenes.
Sistemas de resonancia magnética General Electric (GE) Healthcare
Estos equipos están diseñados para producir imágenes de las estructuras internas del cuerpo y son utilizados para diagnosticar.
La Administración de Drogas y Alimentos de los Estados Unidos (FDA) notificó que se retiraban del mercado por peligro de muerte el 18 de febrero de 2015. Alcanzó a los modelos Discovery MR450, Discovery MR 750, y varias partes de los modelos Signa y Optima.
Cinco modelos Signa ingresaron a la Argentina entre marzo y septiembre de 2015, mientras el alerta seguía vigente. El alerta recién cesó el 10 de septiembre de 2015.
Lo mismo sucedió con distintas partes de los modelos Optima, que entraron en forma continua a la Argentina entre enero y diciembre de 2015.
La ANMAT respondió ante la consulta periodística que la importadora GE Healthcare Argentina “notificó al cliente que hubo un evento adverso. Son equipos de gran volumen. Se cambió un componente”.
Se le trasladó la consulta a General Electric (GE Healthcare). La compañía informó que el recall en referencia a 2015 fue el resultado de un error del usuario y no un defecto del producto. “Para garantizar que este error humano no volviera a suceder en ningún otro lugar, GE Healthcare envió ingenieros de servicio para inspeccionar sus dispositivos en todos los sitios del mundo donde se encontraban instalados”, añadieron. Aseguraron también que esa advertencia fue una acción voluntaria iniciada por GE Healthcare en diciembre de 2014, con el objetivo de poner al tanto a los usuarios y realizar inspecciones globales.
GE Healthcare informó al equipo argentino de ICIJ que, debido a políticas de comunicación global, no es posible compartir datos sobre clientes, cifras y datos de importación.
Sistema de Exploración por Cámara Gamma de General Electric Healthcare
Es un sistema de tomografía computarizada diseñado para procedimientos de imágenes de medicina nuclear general con fines diagnósticos.
La multinacional estadounidense dio cuenta de un incidente en una instalación del Centro Médico VA en los Estados Unidos. Un paciente falleció debido a las lesiones sufridas mientras se lo escaneaba en un Infinia Hawkeye 4, un dispositivo similar.
El 3 de julio de 2013, la compañía notificó a los hospitales que estaba retirando del mercado varios sistemas de diagnóstico por imágenes de medicina nuclear porque podrían producir lesiones graves o muertes debido a una falla en el producto.
La FDA emitió otro alerta para los modelos Brivo NM615 y NM 620 el 24 de julio de 2013. El Brivo NM65 fue importado el 23 de octubre de 2015 GE Sistemas Médicos de Argentina SA, pero el alerta ya había cesado en 2014.
Con relación a este caso, General Electric respondió que “los retiros fueron provocados por un incidente aislado en los EE. UU. Como resultado, se realizó una investigación exhaustiva de los dispositivos en todo el mundo, incluidas todas las unidades en Argentina. Confirmamos que no hubo un riesgo para los pacientes y, por lo tanto, no fue necesario modificar ninguno de los dispositivos en Argentina”.
Infinia Hawkeye 4 Nuclear Medicine System de GE Healthcare
Este fue el modelo que causó la muerte de un paciente en los Estados Unidos. Para cuando las autoridades de ese país emitieron una alerta general el 24 de julio de 2013, la ANMAT ya había autorizado la importación en junio de 2011.
El dispositivo ingresó el 25 de febrero de 2013, importado por Investigaciones Médicas SA. Hay otro ingreso del producto el 11 de abril de 2013, importado por Diagnósticos Maipú por Imágenes SA. Ambas importaciones ocurrieron poco antes de que saltara la alarma. El 7 de octubre de 2014, el Instituto Gamma SA importó una tercera unidad.
Lo llamativo en este caso es que la ANMAT volvió a autorizar la importación del dispositivo en abril de 2014, cuando el alerta seguía vigente en los Estados Unidos (culminó el 19 de junio de 2014). “Hubo acción de campo con un servicio técnico para verificar si había un problema con los equipos a modo de prevención”, informó el organismo de control argentino.
General Electric confirmó que efectivamente, los retiros fueron provocados por una muerte ocurrida en EE. UU y sostuvo que se realizó una investigación de los dispositivos en todo el mundo, incluidas todas las unidades en Argentina. “No hubo un riesgo similar para los pacientes y, por lo tanto, no fue necesario modificar ninguno de los dispositivos en Argentina”.
Discovery NM630 de GE Healthcare
Lo ocurrido con el equipo Infinia hizo que FDA también emitiera un alerta para otro equipo de la misma marca, el Discovery NM 630 de GE Healthcare, por prevención. El alerta sanitaria en los Estados Unidos estuvo vigente entre el 24 de julio de 2013 y el 16 de junio de 2014.
Uno de estos equipos fue importado a la Argentina el 19 de junio de 2013, mientras que otra unidad fue traída al país el 9 de diciembre de 2014, una vez superada la alerta internacional.
Consultada sobre el tema, la importadora Fundación Médica Mar del Plata, reconoció que importó uno de estos equipos NM 630 y dijo que la razón del alerta había sido un equipo diferente, el Infinia. Sin embargo, el alerta había alcanzado al que ingresaron a nuestro país.
Con relación al fabricante, la misma respuesta que General Electric dió para los equipos Brivo e Infinia, fue la que se proporcionó para el Discovery NM 630. Esto es, que se revisaron todos los dispositivos en todo el mundo, incluída la Argentina, y que no hubo riesgo para los pacientes.
Bomba de globos intraaórticos Datascope
Este dispositivo se coloca en la aorta para mejorar el aporte de oxígeno y reducir la carga de trabajo del corazón en casos de infartos. Es fabricado por Datascope y en la Argentina fue importado por la empresa Debene y por la Fundación Favaloro.
El 17 de julio de 2017, la FDA emitió un alerta por peligro de muerte para los modelo CS100 y sugirió verificaciones para el modelo CS300. Este último ya había ingresado a la Argentina siete veces entre marzo y luego diciembre del año pasado. Pero el primer modelo, que implicaba mayor riesgo, también fue importado.
¿Qué sucedió con este dispositivo en la Argentina después del alerta? “La información es confidencial”, contestó la ANMAT cuando el equipo argentino pidió explicaciones sobre la importación de ese dispositivo.
La Fundación Favaloro sí respondió con información a esta consulta. “La Institución cuenta con dos consolas CS100. Fuimos notificados de esto por la empresa Debene el 28 de noviembre de 2017, pero hasta la fecha no han recibido la pieza de reemplazo preventivo”. Los equipos nunca se corrigieron para reparar la posible falla.
“De nuestra parte dejamos indicada la situación en el equipo para que se tomen los recaudos en caso de ocurrencia. Como aclaración, nunca ocurrió esta falla potencial”, agregó la fundación.
Ventilador CareFusion AVEA
Este dispositivo suministra oxígeno al paciente para ayudarlo a respirar.
La FDA emitió un alerta por peligro de muerte el 3 de abril de 2015, que actualmente sigue vigente. El organismo estadounidense anunció que se retiraban del mercado todos los modelos de estos ventiladores de la firma Carefusion. La falla afectaba la válvula de seguridad del dispositivo que hacía que el respirador dejara de funcionar. El paciente, entonces, podía morir.
La ANMAT aprobó a la empresa Driplan SA para que importara este producto en septiembre de 2010, y volvió a autorizarla en febrero de 2013 y el 4 de marzo de 2016, cuando el alerta estaba vigente para todos los modelos.
La importadora trajo el dispositivo seis veces entre marzo y diciembre de 2015; y otras dos veces en febrero de 2016. Luego, en mayo y agosto de 2017, se volvió a importar tres unidades del producto. Las alarmas ya habían saltado, pero el Estado argentino permitió que ingresaran al país.
Driplan SA dio su versión de los hechos: “Los equipos no tenían que ser sacados del mercado, podrían continuar funcionando sin problema. CareFusion lanzó una acción voluntaria correctiva sobre estos equipos sobre un lote y decían que había que reemplazar un componente. Nos mandaron una nota, informamos a ANMAT y procedimos a la acción correctiva que era el cambio del componente”. Consultados sobre qué instituciones médicas tenían los equipos y cuantos equipos fueron distribuidos, señalaron: “Nosotros tenemos un contrato de confidencialidad con la fábrica y no se pueden brindar detalles sobre los equipos en particular”.
La ANMAT informó que “hicieron el cambio de plaquetas del dispositivo” una vez importado y vendido.
Ventilador V60 de Respironics (Philips)
Es un ventilador destinado a mejorar la respiración del paciente.
La FDA emitió alertas el 4 de junio de 2013 y el 24 de abril de 2017 (esta vez sobre algunos modelos y lotes que seguían implicando riesgos). Se registró un ingreso del producto el 25 de noviembre de 2016, importado por Philips Argentina.
La empresa, consultada sobre el particular, manifestó que “en 2013 y 2017 Philips procedió al retiro voluntario del mercado del ventilador Respironics V60.
“En ambos retiros voluntarios, Philips notificó debidamente a clientes, distribuidores y agencias reguladoras de todo el mundo. No se recibió información sobre ningún evento adverso en Argentina”.
Respirador Newport
Newport Medical Instruments realizó un retiro voluntario de ciertas baterías de sus respiradores debido a que podían ocasionar fallas que provocaran la muerte de los pacientes.
El 26 de abril de 2013, la FDA publicó un alerta que duró casi dos años, hasta marzo de 2015. Luego existieron otras alertas nuevas sobre el mismo producto.
El dispositivo ya había ingresado al país, importado por Age Medical SA, en 2010 y 2011. ““Hubo acción de campo”, volvió a limitarse la ANMAT.
La importadora respondió que realizaron el cambio de las baterías de los respiradores distribuidos entre sus clientes a pedido de la representante local del fabricante, Covidien, que ahora forma parte de Medtronic.
Sistema de localización Intraoperatorio BrainLab
Este dispositivo permite a los cirujanos hacer incisiones en el cráneo del paciente con mayor precisión. Requiere que el médico ingrese datos de manera digital. Pero en 2016, se informó en los Estados Unidos que un error en el software del dispositivo “podría intensificar pequeñas imprecisiones” en las incisiones del cráneo de los pacientes.
La FDA emitió un alerta el 19 de enero de 2016, que duró más de dos años. Dos de estos dispositivos fueron importados por AADEE SA luego del alerta, el 20 de noviembre de 2016. “Hubo acción de campo. Se hizo una actualización del software”, justificó la ANMAT.
Desde AADEE no respondieron en forma particular y sólo señalaron: "En términos generales cuando el proveedor avisa que hay un problema con el software se le avisa a los clientes. La casa matriz nos envía el nuevo software y los técnicos corrigen los equipos".
Sistema de Asistencia Ventricular HeartWare
Este equipo es una bomba mecánica que sustituye a las bombas naturales del corazón cuando éstas fallan. Está indicado para pacientes con insuficiencia cardíaca.
Entre 2013 y 2018, hubo al menos 13 alertas de la FDA por peligro de muerte. Durante esos años, el producto ingresó al país importado por Medikar SA. “Hubo un problema con el pin, que se podía doblar. La acción de campo es advertir que de no ser colocado correctamente el pin se podía doblar. Se reforzaron las instrucciones de uso”, admitió la ANMAT.
Sin embargo, la importadora asegura que si bien ingresó al país seis unidades del dispositivo “los números de serie no están comprendidos en la alerta”. “Se han importado 6 dispositivos correspondientes al Modelo 1407AR, y ninguno de ellos se corresponde con el rango de números de serie involucrados. Del total de 6 dispositivos importados, 4 fueron utilizados y 2 permanecen en stock en la empresa”, agregaron. “El fabricante siempre que corresponde nos informa. Respecto a esta alarma en particular aún no hemos recibido comunicación, entendemos se debe a que no nos involucra”.
Balón o globo de contrapulsación aórtica Arrow
Es un dispositivo utilizado en pacientes que han sufrido un infarto agudo de miocardio.
Las autoridades estadounidenses emitieron alertas para los modelos “Arrow IAB de 30 cc” en 2014 y 2016, que sigue vigente hasta la actualidad. “El cuerpo del dispositivo puede separarse del eje. Si se produce la separación, existe la posibilidad de sangrado”, marca el comunicado.
El producto es fabricado por Arrow International, una división de Teleflex Medical.
Entre abril y noviembre de 2015 ya habían ingresado al país 7 modelos de estos globos. Otras 16 unidades ingresaron mientras regía el alerta (7 entraron entre junio y diciembre de 2016 y nueve más fueron importados entre febrero y noviembre de 2017).
Según la ANMAT, la importadora, American Fiure SA, informó el evento. “No coinciden los lotes que tuvieron el problema”, aclararon. Pero la importadora desmintió al organismo.
American Fiure SA respondió que “efectivamente, en el año 2016 se llevó a cabo una acción de campo. Retiramos del mercado las unidades que habíamos distribuido hasta el momento y las despachamos de vuelta a la fábrica de origen. No tuvimos reportes de eventos con pacientes, entendemos que todos los productos que distribuimos fueron recuperados y devueltos al fabricante”.
Un sistema que mira al norte equivocado. Cuando el equipo argentino de ICIJ consultó a la ANMAT porqué no había alertado a la población cada vez que estos dispositivos médicos registraron fallas que podrían poner en riesgo la vida de los pacientes, el organismo sostuvo que sólo emite resoluciones públicas “cuando hay una amenaza seria a la salud pública”. Si son “casos muy puntuales” no se publica en Boletín Oficial “para no alarmar a la población”.
La información sobre estos eventos es “confidencial”, sostiene el organismo. La información queda entre la empresa y el organismo de control. El fabricante decide cuál es la acción correctiva porque conoce el producto, afirman. “Se analizan los riesgos y el costo-beneficio: si algo está implantado en el cuerpo, por ejemplo, puede ser más riesgoso sacarlo que dejarlo”, resumen.
La ANMAT explicó que los importadores atraviesan una inspección cada tres años para poder renovar el Certificado de Buenas Prácticas de Fabricación (BPF), que especifica cómo debe llevar a cabo su trabajo, cómo debe reportar un evento adverso y cómo realizar las acciones correctivas de los productos con alertas.
“Nosotros llegamos hasta la puerta, nos cercioramos que el producto llegue en buenas condiciones y bien conservado a la institución para una intervención médica. Luego en la práctica médica en sí, no nos metemos”, afirman.
(*) El equipo argentino del Consorcio Internacional de Periodismo de Investigación que participó de The Implant Files está integrado por Emilia Delfino (PERFIL) Mariel Fitz Patrick (Infobae); Sandra Crucianelli (para Perfil); Maia Jastreblansky, Iván Ruiz y Ricardo Bron (La Nación).







