

Pueden salvarnos la vida. O destruirla. Pueden aliviar el dolor o agudizarlo. Pueden ser pequeños o enormes. De silicona o de titanio. Lo que sí es común a todos los dispositivos médicos es que mueven miles de millones de dólares al año pero nadie los controla.
En esta nueva investigación global denominada “The Implants Files”, el Consorcio Internacional de Periodistas de Investigación (ICIJ por sus siglas en inglés) expone a la industria médica y cómo funciona el negocio de los dispositivos médicos. Prótesis de rodilla y cadera, marcapasos, implantes mamarios, pequeños e inmensos artefactos tecnológicos, desde bombas de insulina hasta desfibriladores y tomógrafos, que pueden salvarnos o arruinarnos.
Esta es la primera entrega de una serie de notas que confirmarán el rol que juegan médicos, obras sociales, las prepagas y el Estado frente a una industria que tiene el control casi absoluto de los dispositivos que vende. Una industria que ha demostrado que sabe jugar a quebrar las reglas y recurrir a prácticas corruptas para asegurar sus ventas y defraudar al Estado. Cirugías que nunca sucedieron, prótesis facturadas al Estado que nunca fueron implantadas, médicos que reciben sobornos para favorecer a un fabricante.

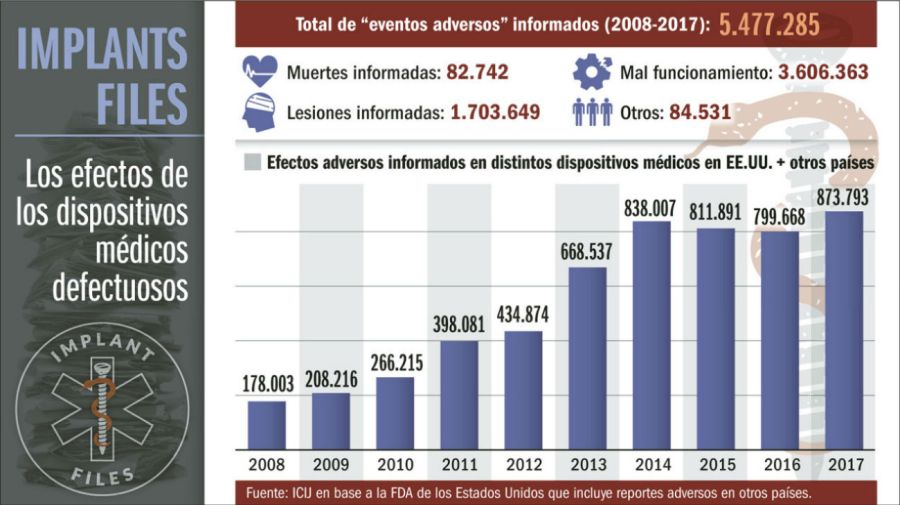
Tanto en Estados Unidos con en el resto del mundo, entre 2008 y 2017, se reportaron 5.477.285 de fallas en distintos dispositivos médicos de algunas de las principales compañías a nivel mundial. De dichos reportes adversos, 82.742 se vincularon a la muerte de pacientes que recibieron tratamiento con dispositivos médicos supuestamente probados y seguros. Otro 1,7 millón de personas resultaron con lesiones o problemas de salud. Así lo determinaron las cifras obtenidas por ICIJ tras el análisis de información pública de la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA).
El equipo argentino, integrado por La Nación, Infobae y PERFIL, entrevistó a funcionarios y exfuncionarios del área de Salud, a especialistas, sindicalistas, empresarios y a pacientes damnificados que siguen de cerca a esta industria, integrada por grandes compañías multinacionales y por fabricantes e importadores nacionales que por primera vez son investigados de forma global. Además, detectó 13 episodios en los cuales grandes compañías extranjeras debieron retirar sus productos del mercado argentino porque se detectaron fallas con peligro de muerte para los pacientes, pero éstos tal vez nunca lo supieron.
Algunos de los productos incluso ingresaron al país después de que habían sido retirados del mercado en los Estados Unidos. En al menos cuatro casos, ingresaron cuando la alerta estaba vigente.

Para esta investigación, ICIJ creó una base de datos de acceso público que, en primera instancia, permite explorar más de 70.000 retiros de dispositivos médicos, alertas y avisos de seguridad de más de 11 países, para que los pacientes en todo el mundo a partir de ahora tengan acceso a datos sobre dispositivos médicos defectuosos o peligrosos, incluidos los que ponen en peligro la vida.
Consultá acá la base de datos online sobre dispositivos médicos defectuosos
El capítulo local demuestra que en Argentina el sistema no puede evitar fallarle dos veces a los pacientes: en primer lugar, al arriesgar que se instalen en sus cuerpos dispositivos médicos defectuosos, que pueden ocasionar graves daños a la salud de las personas, e incluso la muerte; y en segundo lugar, al orquestar un circuito de alertas que rara vez llega al paciente comprometido. Los argentinos pueden pasarse una vida sin conocer que llevan dentro de sus cuerpos dispositivos defectuoso que ponen en riesgo su vida. Esa ignorancia se termina cuando el dispositivo falla. Muchas veces puede ser demasiado tarde.
Cuando fallan. Este sistema no es sólo “made in Argentina”. Lo mismo sucede en los Estados Unidos y en países de Europa y de América Latina, con distintos niveles de gravedad.
La investigación en distintos lugares del mundo demuestra que en casi todas las jurisdicciones se repite el mismo procedimiento. Los fabricantes de estos dispositivos son quienes prueban y garantizan la supuesta seguridad del producto y las autoridades estatales toman esa información como válida sin casi ningún tipo de control. Cuando un dispositivo falla y debe ser retirado del mercado es el fabricante el que debe informar a los gobiernos sobre la falla y encargarse de sacar al producto de circulación.
En Argentina, el organismo que debe controlar a estas empresas es la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica, bajo la órbita del Ministerio de Salud. Su misión es garantizar “que los productos para la salud sean eficaces, seguros y de calidad”.
La ANMAT se ha hecho popular por sus alertas sobre productos de consumo masivo que el organismo anuncia que deben ser retirados del mercado. En los medios y en el Boletín Oficial, suele informar a la población qué alimentos, insecticidas, artículos de limpieza y de cosmética no deben ser consumidos porque implican un riesgo para la salud o porque no cumplen con la reglamentación. Pero cuando entran en juego las grandes compañías de dispositivos médicos el procedimiento es diferente: la comunicación no es pública, es decir, no llega al paciente. El circuito es cerrado para la comunidad médica y según las autoridades del organismo “es confidencial”. La ANMAT sostiene que informa a los hospitales e instituciones médicas y que la comunicación a los pacientes afectados queda a merced de los médicos. Los mismos médicos que al revelar la verdad a sus pacientes pondrían en tela de juicio su propia praxis.
El secretario de Salud, Adolfo Rubinstein, reconoce las debilidades del sistema. “La responsabilidad del Estado es informar a la ciudadanía y emitir un alerta que llegue a las importadoras, a las instituciones sanitarias y a los médicos para que retiren un determinado producto del mercado. Pero la acción de retiro la hace la empresa importadora o la institución médica. Que el retiro lo haga el Estado es imposible, acá y en el mundo. La responsabilidad de contactar a los pacientes o de no usar más un dispositivo es de la institución o del profesional médico”.
Rubinstein, máxima autoridad sanitaria del país, también coincide en que el Estado no puede controlar si las compañías cumplen o no con las reglas cuando deben retirar un producto defectuoso del mercado. “Cuando la ANMAT recibe una denuncia, hace una evaluación de los riesgos y emite un alerta. Pero no tiene capacidad de hacer un seguimiento de qué pasó con cada producto médico”, asegura.

¿Cuánto vale una vida para la industria médica? En los tribunales porteños aplican un cálculo inmobiliario, explica un protagonista del día a día en los juicios de pacientes contra médicos e instituciones. “Una muerte en Capital Federal vale un departamento de tres ambientes en Caballito”. La incapacidad generada a un paciente por un daño físico equivale a US$ 1.000 por cada punto de incapacidad. Esto no está reglamentado ni normado.
Virginia Luna sabe lo que significa lidiar con este sistema. Esta abogada representa a 1500 argentinas que se colocaron prótesis mamarias de la marca francesa Poly Implant Prothèse (PIP) y tiempo después descubrieron que lo que llevaban dentro de su cuerpo podía estallar porque el fabricante había utilizado un gel de silicona de mala calidad y bajo costo que ni siquiera estaba autorizado.
El caso de los implantes PIP demuestra que el mercado de dispositivos médicos surfea entre lagunas de vacío jurídico y un Estado que no toma la decisión de controlar qué tipo de materiales se introducen en los cuerpos de las personas. Una vez más, la salud de los pacientes quedó a merced de los fabricantes e importadores de siliconas.
Las clientas de Luna demandaron a la empresa francesa que fabricó y vendió prótesis PIP. También decidieron demandar a la entidad certificadora alemana TÜV Rheinland, que permitió que el producto saliera al mercado.
Las PIP tenían una mayor probabilidad de romperse dentro del cuerpo de las pacientes. El escándalo derivó en que el fundador de PIP, Jean-Claude Mas, fuera condenado en Francia en mayo de 2016 a cuatro años de cárcel. Su empresa utilizó un gel no autorizado durante una década. Fue acusado de fraude y estafa. Su empresa quebró. Como muchas de sus siliconas. El caso se hizo público en 2010 pero aún hay damnificadas que esperan recibir una indemnización de la fabricante francesa.
Las PIP son un claro ejemplo de cómo América Latina puede convertirse en el patio trasero de los dispositivos médicos del primer mundo. Se fabricaron 500.000 prótesis defectuosas y 400.000 tuvieron como destino a Latinoamérica. Sólo 100.000 se vendieron en Europa y el resto del mundo. Unas 15.000 se vendieron a la Argentina.
Ante el escándalo desatado en Europa y la catarata de demandas contra el fabricante y los cirujanos, el Estado argentino no pudo hacer nada. Cada dispositivo médico, sea una prótesis o una silicona, lleva un sticker con un número de serie que lo identifica y permite su rastreo. Pero la ANMAT ni siquiera tenía un mecanismo para rastrearlos e identificar a las pacientes en peligro porque no contaba con la trazabilidad de los implantes.
Recién después de este caso y tras varias presentaciones de la abogada Luna, la ANMAT cambió su normativa y comenzó a trazar la ruta de los implantes.
Este caso también expone otra debilidad más del sistema, que persiste aún en la actualidad. El Estado -a través de la ANMAT- no obliga a los médicos a reportar a sus pacientes que han sido operados o tratados con un dispositivo médico defectuoso, ya sea una prótesis, un marcapasos o implantes de mamas. Luna dice que el organismo “sugiere” a los médicos informen sobre los descubrimientos negativos y riesgos que salen a la luz sobre estos dispositivos pero no hay ninguna garantía de que los cirujanos acaten la “sugerencia”. La salud del paciente queda nuevamente a merced de un tercero involucrado: su médico.
El exministro de Salud de Mauricio Macri, Jorge Lemus, intentó que el Congreso sancione una ley para crear un organismo nuevo que, sostiene, ayudaría a fortalecer los derechos del paciente frente al sistema. Lemus propuso crear la Agencia Nacional de Evaluación de Tecnologías de Salud (AGNET)
En su oficina de la Academia Nacional de Medicina, Lemus explica que Argentina dilata desde hace décadas la creación de este organismo que, de cumplir su función con transparencia, podría ayudar a controlar el uso de dispositivos médicos y a contener el lobby de sus fabricantes, los negociados con el Estado y la corrupción médica. Este es otro punto que abordará la investigación global de The Implants Files. ¿Qué hace el Estado para controlar la corrupción médica? ¿Cómo funciona el submundo de los cirujanos que reciben dinero de los fabricantes de dispositivos médicos para favorecer sus ventas? El equipo argentino de ICIJ identificó al menos cuatro casos que explican estos mecanismos y tres de ellos están impunes en el país.
Mecanismo argentino. La ANMAT tiene la misión de aprobar los dispositivos para su comercialización en el país, tanto los de origen nacional como extranjero. Para clasificar el riesgo potencial de los dispositivos utiliza un índice que va de Clase I a Clase IV, siendo este último el de mayor riesgo: muerte. Este criterio es inverso al que utiliza la FDA.
Para esta organización, los Clase I son aquellos peligrosos o defectuosos que podrían causar problemas de salud graves o la muerte. Los Clase II son los que pueden causar un problema de salud temporal o que representan una amenaza leve, mientras que los Clase III son los que probablemente no causen reacciones adversas a la salud, pero violan las leyes de fabricación o etiquetado de la FDA.
¿Quién decide qué tan riesgoso es un dispositivo médico? “La empresa importadora del producto, pero la ANMAT corrobora que sea correcto”, explican en el organismo. ¿En base a qué pueden corroborar o desestimar el riesgo informado por los dueños del producto? “En base a la documentación que entrega el importador”, contestan.
La ruta de un dispositivo en la Argentina no tiene muchas escalas. Los requisitos que la ANMAT exige a las compañías importadoras son los mismos que exige a los fabricantes nacionales de dispositivos:
1) disponer de un depósito para el almacenamiento de los productos.
2) registrar los productos que quiere importar.
3) obtener un certificado de buenas prácticas que emite la propia ANMAT.
El Estado recibe del importador la información del producto que envía el fabricante, que incluye la información técnica y el manual de uso del dispositivo médico. En ciertas ocasiones, también Se solicitan copia de los ensayos realizados por el fabricante para probar su producto. El fabricante decide el índice de riesgo. La ANMAT lo corrobora. El dispositivo está listo para ingresar al país.
¿El Estado no hace algún tipo de ensayo sobre los dispositivos médicos? “No, el Estado no tiene la capacidad de hacer pruebas sobre los dispositivos, se toman por buenos los enviados por el fabricante”, explican en el organismo.
Un sistema con más sombras que luces. Charles Rosen es un cirujano especialista en columna vertebral y comprometido con la seguridad de los pacientes. En una entrevista con ICIJ describió cuál es el modelo de este mercado: desarrollar un producto. Sacarlo rápido. Ocultar las complicaciones. Pagar millones a consultores por estudios favorables. Pagar documentos para usar el dispositivo en pacientes que obtienen poco o ningún beneficio de ellos. "La compañía gana miles de millones", dice Rosen.
Héctor Villalba es un experimentado médico legista que estudia casos de mala praxis desde hace décadas. “Para la provisión de cualquier prótesis o implante, las coberturas sociales o prepagas trabajan con determinados distribuidores. Asimismo, los médicos también suelen requerir que la prótesis prescrita sea adquirida a determinado proveedor; en ambos casos porque suelen tener algún beneficio económico. Esto explica que muchas veces el paciente queda en medio de una disputa entre el médico y su cobertura social, porque cada uno quiere imponer a su proveedor”, explica en una entrevista. Por eso, agrega Villalba, “en estos casos de dispositivos médicos, como prótesis o marcapasos, en lo que respecta a mala praxis, la pregunta que hay que hacerse es si era necesario colocarlo o no, si están bien indicados o no, si se ocasionó un perjuicio o no al implantarlos”.
Marcelo Peretta es doctor en Farmacia y Bioquímica y es Secretario General del Sindicato de Farmacéuticos y Bioquímicos. Los directores técnicos de las empresas importadoras de estas prótesis e implantes suelen estar afiliados a su gremio. La ANMAT autoriza a estas compañías a importar los dispositivos luego de un procedimiento estándar.
“Los organismos de control están cooptados por la industria, tanto en el caso de la FDA de los Estados Unidos como en el caso de la ANMAT”, afirma Peretta. “El problema es que el paciente no lo sabe y cuando hay fallas se ocultan. Hay un ocultamiento deliberado de los efectos adversos.
Los dispositivos médicos -a diferencia de los medicamentos- se aprueban por similitud, como innovación de otro producto anterior ya aprobado, y los efectos adversos se ven cuando están ya colocados en los pacientes”, agrega.






